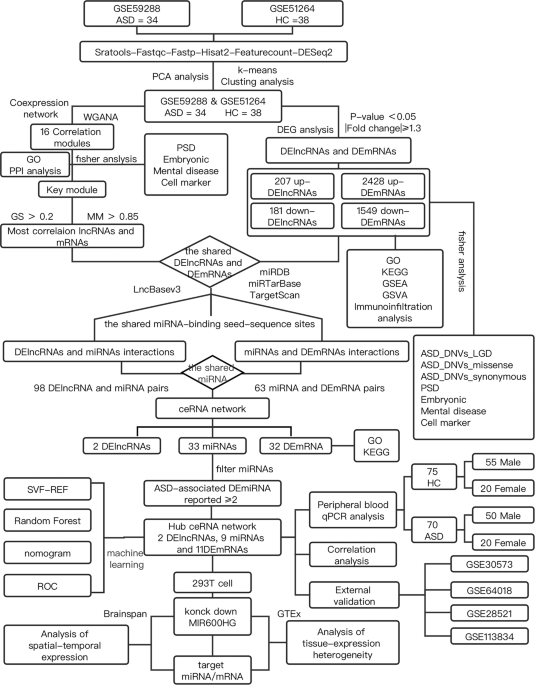
Abstract Autism spectrum disorder (ASD) is a complex disorder of neurodevelopment, the function of long noncoding RNA (lncRNA) in ASD remains essentially unknown. In the present study, gene networks were used to explore the ASD disease mechanisms integrating multiple data types (for example, RNA expression, whole-exome sequencing signals, weighted gene co-expression network analysis, and protein-protein interaction) and datasets (five human postmortem datasets). A total of 388 lncRNAs and five co-expression modules were found to be altered in ASD. The downregulated co-expression M4 module was significantly correlated with ASD, enriched with autism susceptibility genes and synaptic signaling. Integrating lncRNAs from the M4 module and microRNA (miRNA) dysregulation data from the literature identified competing endogenous RNA (ceRNA) network. We identified the downregulated mRNAs that interact with miRNAs by the miRTarBase, miRDB, and TargetScan databases. Our analysis reveals that MIR600H G was downregulated in multiple brain tissue datasets and was closely associated with 9 autism-susceptible miRNAs in the ceRNA network. MIR600HG and target mRNAs ( EPHA4 , MOAP1 , MAP3K9 , STXBP1 , PRKCE , and SCAMP5 ) were downregulated in the peripheral blood by quantitative reverse transcription polymerase chain reaction analysis (false discovery rate <0.05). Subsequently, we assessed the role of lncRNA dysregulation in altered mRNA levels. Experimental verification showed that some synapse-associated mRNAs were downregulated after the MIR600HG knockdown. BrainSpan project showed that the expression patterns of MIR600HG (primate-specific lncRNA) and synapse-associated mRNA were similar in different human brain regions and at different stages of development. A combination of support vector machine and random forest machine learning algorithms retrieved the marker gene for ASD in the ceRNA network, and the area under the curve of the diagnostic nomogram was 0.851. In conclusion, dysregulation of MIR600HG , a novel specific lncRNA associated with ASD, is responsible for the ASD-associated miRNA-mRNA axes, thereby potentially regulating synaptogenesis. Introduction Autism spectrum disorder (ASD) refers to a group of early-onset, lifelong, clinically heterogeneous, neurodevelopmental disorders with deficits in social functioning and the presence of repetitive and restricted behaviors or interests [1]. ASD also manifests significant genetic heterogeneity; thousands of common variants and rare, de novo single nucleotide mutations are estimated to contribute to ASD [1, 2]. Some studies have shown that ASD-associated mutations affect both coding and noncoding parts of the genome [3]. Most annotation sites in the human genome are noncoding, and a significant portion of noncoding transcripts are represented by long noncoding RNAs (lncRNAs) [4], which are defined as RNA molecules with >200 nucleotides. Among all other ncRNAs, lncRNAs are highly expressed in the human brain-specific regions of the neural tissues [5]. The lncRNAs are also involved in brain development and neurogenesis; thus several lncRNAs lead to defective neurogenesis and abnormal neural circuits after knockdown or aberrant alternative splicing [6, 7]. However, the contribution of the regulatory mechanisms of these lncRNAs with respect to ASD is yet to be elucidated. Recent studies have identified the contribution of genetic, environmental, epigenetic, neuropathological, and immunological factors [8, 9]. LncRNAs play a role in various biological processes, including epigenetic regulation, chromatin remodeling, and the regulation of transcription and translation levels [10]. Several studies have shown that lncRNA is involved in many key biological functions and responds to environmental factors in the brain [3]. lncRNAs are fundamental regulators of transcription and can regulate susceptibility genes involved in psychiatric disorders and neurodevelopment [11,12,13]. The disruptions in lncRNAs, such as SHANK2-AS and BDNF-AS, can affect synapse, neuron function, and the development of autism [14]. Hitherto, only a small fraction of lncRNAs in the brain has been studied. For example, two primate-specific lncRNAs, LINC00693 and LINC00689, are upregulated in the ASD cortex [11]. The competing endogenous RNA (ceRNA) theory proposes crosstalks between ncRNAs and coding RNAs via microRNA recognition elements (MREs), which are microRNA (miRNA) complementary sequences [15]. However, the specific role of ceRNA networks in ASD has not been elucidated. In the present study, weighted gene co-expression network analysis (WGCNA) and differential analysis were performed to screen the disease-associated lncRNAs, while integrating multiple data sources (such as five human postmortem datasets, GTEx, and Brainspan) to probe into ASD disease mechanisms using gene networks. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) assays confirmed the dysregulation of lncRNAs and target genes at the peripheral circulation level. The in vitro experiments characterized MIR600HG for regulating the synapse-related genes through the ceRNA mechanism. Herein, our comprehensive bioinformatics analysis provides a framework for assessing the functional participation of lncRNAs in ASD. Materials and methods Data collection and differential expression analysis The RNA-seq data of the prefrontal cortex from 34 autism patients in the GSE59288 and 38 normal samples in the GSE51264 dataset [16], obtained by sequencing on the Illumina HiSeq 2000 GPL11154 platform, was collected from the NCBI Gene Expression Omnibus (GEO) database. The microarrays and RNA-seq datasets, including GSE30573 (high coverage RNA-seq), GSE64018 (high coverage RNA-seq), GSE113834 (expression microarray), and GSE28521 (expression microarray), used for validation were integrated (Supplementary Table S1). First, the downloaded SRA file contained the sequencing reads for each sample, and its quality control was performed by FastQC (version 0.11.5). Then, the filtered reads were used to map to the hg38 genome reference genome (http://ftp.ensembl.org/pub/release-104/gtf/) using HISAT2 (version 2.1.0) [17] with default parameters. Third, the bam file was quantified using featureCounts [18] and filtered to remove the low-count genes expressed in <75% of samples. Finally, based on the raw counts matrix, we identified the differentially expressed protein-coding RNAs (DEmRNAs) and differentially expressed lncRNAs (DElncRNAs) using DEseq2 (|fold-change (FC)| > 1.3 and p -value < 0.05) for further analysis. Functional and pathway enrichment analyses Gene ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, gene set enrichment analysis (GSEA), and gene set variation analysis (GSVA) were performed using the R package “clusterProfiler” [19]. The cutoff criterion was p < 0.05. The datasets of multiple psychiatric disorders [20], the gene set of rare de novo variants associated with ASD by whole-exome sequencing study (WES) [21], different types of neuronal cell markers [22, 23], postsynaptic density [24], and embryonic development [21] were integrated for GSEA using Fisher’s exact test. The details of gene set selection and sources are summarized in Supplementary Table S2, p -values were adjusted for multiple comparisons using Benjamini–Hochberg correction to assess the false discovery rate (FDR). Screening of key modules based on WGCNA Weighted gene co-expression network analysis facilitates the classification of genes based on their similar expression patterns. We used the “WGCNA” R package [25] to construct a scale-free co-expression network that adheres to the scale-free property. The scale-free network exhibits a power law distribution, which closely resembles the biological reality and demonstrates robustness. The soft-threshold power β = 6 was selected to construct a scale-free network. The adjacency matrix was transformed into a topological overlap matrix to describe the similarity between nodes. Furthermore, module-trait correlations were calculated to screen for modules with a significant correlation with autism ( p < 0.05). The GSEA of each module was carried out using Fisher’s exact test. We extracted a protein-protein interaction (PPI) subnetwork for each associated module from the STRING database [26]. WGCNA was used to identify modules and susceptibility genes associated with autism. In this study, the corresponding gene significance (GS) and module membership (MM) of each gene in the core module were estimated. The genes satisfying the screening criteria (|MM| > 0.85 and |GS| > 0.2) were selected for further analysis. Pearson’s correlation coefficient was used to calculate the correlations between genes. Construction of ceRNA network In order to identify candidate genes for the ceRNA network, the most significant lncRNAs and mRNAs based on WGCNA were intersected with the DElncRNAs and DEmRNAs. The differentially expressed candidate lncRNAs were input into the online database lncbaseV3 (https://diana.e-ce.uth.gr/lncbasev3) to identify the putative binding to miRNAs. We also identified the targeted miRNAs that interact with candidate mRNAs based on interactions generated by the miRTarBase [27], miRDB [28], and TargetScan [29] databases. Next, we searched for case-control studies exploring the differentially expressed miRNAs (DEmiRNAs) between patients with autism and healthy controls from multiple documents [30]. The interactions between DEmiRNAs and lncRNAs or mRNAs were integrated to construct a hub ceRNA regulatory network. Expression pattern analysis of ceRNA network Pearson’s correlation analysis was used to determine any positive correlations between DEmRNAs and DElncRNAs. The tissue-expression heterogeneity of DEmRNAs and DElncRNAs in the ceRNA network was explored using the Genotype-Tissue Expression database (GTEx) (https://www.gtexportal.org/) and FUMA GWAS (https://fuma.ctglab.nl) [31]. Further, we retrieved spatial and temporal expression of the transcriptome in the human brain generated from the BrainSpan project (http://www.brainspan.org/). Participants This study enrolled 70 Han Chinese autistic children (male/female ratio: 50/20) with a mean age of 3.68 (±0.61) (±standard deviation) years. A total of 75 age- and sex-matched healthy children (male/female ratio: 55/20) from the same ethnic group were selected as controls. All participants were recruited at the Peking University Sixth Hospital (Beijing, China). The peripheral blood samples were collected from all participants. Individuals with ASD were diagnosed based on DSM-IV criteria and had no other neuropsychiatric, metabolic, or immune-related conditions. Quantitative reverse transcription polymerase chain reaction The RNA was extracted using the Tiangen RNAsimple Total RNA Kit (Tiangen, DP419, Beijing, China) and reverse-transcribed using a FastKing reverse transcriptase kit (Tiangen, KR116-02, Beijing, China), TransGen TransScript miRNA First-Strand cDNA Synthesis SuperMIX (TransGen, Beijing, China), and lnRcute lncRNA First Strand cDNA Synthesis Kit (Tiangen, KR202-02, Beijing, China). qRT-PCR was performed on a LightCycler 96 (Roche, Switzerland). The expression of the target lncRNA and mRNAs was normalized to that of the control GAPDH . For miRNAs, data were normalized with endogenous control RNU6 . All primers are listed in Supplementary Table S3. For the robustness of the results, genes with low gene expression abundance (CT value > 32) were excluded. The relative quantitation for genes was calculated using the 2 −∆∆Ct method. Cell culture The human embryonic kidney (HEK) 293 cells were obtained from the American Type Culture Collection (ATCC) and regularly checked for mycoplasma contamination. For cell culture, the cells were maintained in high-glucose Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS, GIBCO, 10099-141C, USA). For plasmid transfection, the cells were inoculated into 6-well plates and the plasmids were infected into 293 cells at 70% confluence. The expression plasmid [pLKO.1-CMV-copGFP-PURO or pLKO.1-sh-MIR600HG-CMV-copGFP-PURO] (Supplementary Table S3) was transfected using the jetPRIME® transfection reagent, following the manufacturer’s instructions. After 6 h, the medium was changed to fresh DMEM containing 10% FBS and cultured for 24 h. Diagnostic nomogram for ASD based on machine learning algorithms A combination of support vector machine (SVM) recursive feature elimination (SVM-RFE) algorithm and random forest (RF) were used to screen the potential genes in the ceRNA network using the “e1071,” “randomForest” R package [32]. Next, a diagnostic nomogram was established based on the overlapping genes generated by SVM-RFE and RF algorithms. The nomograms were generated via the R package “rms”. Finally, the receiver operating characteristic (ROC) curve was used to investigate the efficiency of this diagnostic model. The area under the curve (AUC) > 0.65 was considered significant. Statistical analyses All data were analyzed using SPSS. The differences in the relative expression for each mRNA and lncRNA between patients with autism and controls were examined using a nonparametric Mann–Whitney U test (two-tailed). Data are presented as the median and interquartile range. FDR was used for multiple comparison corrections. Downregulation of lncRNA and corresponding miRNA and mRNA regulatory axes was verified in cell experiments, and statistical significance was calculated using unpaired Student’s t -test (two-tailed). All data in accordance with the normal distribution were represented as means ± SEM. The threshold of significance accepted for all statistical analyses was the p -value or FDR < 0.05. Results DEmRNAs and DElncRNAs Identification We profiled differential gene expression (DGE) analysis in 72 postmortem brain tissue samples from 34 ASD cases (GSE59288) and 38 controls (CTL) (GSE51264). A filtering flowchart for the study is shown in Fig. 1. The 388 DElncRNAs (207 upregulated and 181 downregulated) were identified between CTL and ASD samples. We also identified 2428 up- and 1549 downregulated DEmRNAs (Fig. 2 and Supplementary Table S4). Principal component analysis (PCA) revealed the following findings (Fig. 2C): the ASD samples were separated from CTL samples by PCA1. Enrichment analysis (Fisher’s exact test) for gene sets of interest To explore the correlation between the DEmRNAs and the neural cell type marker gene sets, multiple psychiatric disorders, and broad scope of ASD risk genes, including genes with evidence of de novo risk variants, we systematically assessed whether the DEmRNAs are enriched for these genes (Fisher’s exact test, FDR < 0.05; “Methods” section). We observed that the DEmRNAs are significantly enrichment for a variety of neural cell type marker genes. The ASD-upregulated genes are associated with oligodendrocyte, astrocyte and microglial function, while the downregulated genes are associated with neurons, CA1 pyramidal neurons, S1 pyramidal neurons, interneurons and mural cells (Fig. 2E and Supplementary Table S2). The other types of immune cell marker analysis found that CD4+ memory T cells were enriched in the upregulated genes (Supplementary Fig. S1). Interestingly, the downregulated DEmRNAs are enriched for genes causally connected with autism (Fig. 2F) but not for other brain disease-associated genes. We also observed enrichment for the downregulated DEmRNAs hit by de novo variations (likely gene-disruptive (LGD) mutations, missense, and synonymous). These downregulated DEmRNAs also significantly overlapped with encoding postsynaptic density (PSD) proteins and embryonically expressed genes (Fig. 2G and Supplementary Table S2). Pathway enrichment analyses The hierarchical clustering of biological process (BP) terms by measuring similarity revealed that the upregulated set was related to immunity and cell adhesion function (Fig. 3A). The downregulated set was divided into several categories, including “regulation of axonogenesis,” “nervous development,” and “synaptic function” (Fig. 3B). Next, we performed a series of pathway enrichment analyses to characterize these genes. The NOD-like receptor signaling pathway and the PI3K-Akt signaling pathway are two immune-related processes that are linked to the upregulated DEmRNAs (Fig. 3C). Notably, the downregulated groups are involved in the activation of synaptic-related pathways (such as GABAergic synapses and glutamatergic synapses), Calcium signaling pathways, and oxytocin signaling pathways; these pathway targets form a network of tighter interactions than accidentally expected, providing independent confirmation of pathway-level co-regulation (Fig. 3D). Based on these findings, we defined the changing trend of molecular pathways and conducted GSEA analysis on the two groups of samples (Fig. 3E, F). The results of the GSEA analysis on samples shared similarities with those of GO and KEGG analyses of the DEGs and also exhibited specific characteristics. Interestingly, the intestinal immune network for the IgA production pathway is upregulated in ASD (Fig. 3E). The details of GSEA pathways are described in Supplementary Fig. S2. The results of the GSVA analysis (Fig. 3G) further confirmed the reliability of the GSEA results. First, the pathways related to the inflammation immune microenvironment-related pathways (RIG-I-like receptor signaling pathway and NOD-like receptor signaling pathway) were significantly upregulated in the ASD group, as well as the metabolic pathways of fatty acids. In line with the desired results, the downregulated pathways in the ASD group were largely consistent with the results of GSEA, which mainly focused on the pathways related to axon development, neuron projection guidance, and regulation of synapse structure or activity (Fig. 3F, G). Perturbation of lncRNA co-expression modules in ASD brain To further assess the correlations between lncRNA expression changes and disease status, we applied WGCNA (Supplementary Fig. S3 and Supplementary Table S5) to assign the lncRNAs and mRNAs to co-expression modules. Subsequently, 16 modules (Fig. 4A) were identified, and their biological function was examined. Next, we identified five modules that were correlated with the disease status (Pearson’s correlation, p < 0.05); three downregulated (M1, M3, and M4) and two upregulated (M8 and M10) in ASD samples (Supplementary Fig. S3F–J). The GSEA (Fisher’s exact test) of each module (Supplementary Table S6) showed that the upregulated M8 module was associated with glial cell differentiation and enriched in oligodendrocytes. On the other hand, the M3 (nervous system development) and M4 (synaptic signaling) modules showed highly significant enrichment for known autism susceptibility genes and multi-neuronal markers. The M3 modules showed significant correlations with age and hence, were excluded from subsequent analysis. However, M4 was significantly correlated with disease status but not correlated with critical experimental covariates (age and sex) (Fig. 4A). Next, we plotted the PPI network of the modules to show functional interactions between proteins, with the strongest protein interactions for the M4 module (Fig. 4B–F). The key gene co-expression module (M4) was significantly correlated with ASD, enriched for DEGs between ASD and control (Supplementary Table S4). The screening criteria for the DElncRNAs and DEmRNAs of the M4 module were based on the MM and GS assessment (Supplementary Table S7; “Methods”). Identification of the ceRNA network associated with ASD We first searched for the target miRNAs of the DElncRNAs from the M4 modules and identified 98 lncRNA-miRNA interactions, as described above. Next, the potential interactions of 63 miRNAs with DEmRNA, based on a collection of interactions supported simultaneously by three miRNA target prediction databases, were identified. A preliminary ceRNA regulatory network exhibited a positive correlation between the expression of lncRNA and mRNA (Fig. 5A; Supplementary Table S8). Next, we searched for case-control studies on the miRNA dysregulation between patients with autism and healthy controls from multiple documents. A total of 74 ASD-associated DEmiRNAs, reported from at least two previous case-control studies (Supplementary Table S9), were selected. The lncRNA-miRNA-mRNA associations with the DEmiRNA as the core were screened, and a hub ceRNA network consisting of 2 DElncRNAs, 9 DEmiRNAs, and 11 DEmRNAs was constructed (Fig. 5B; Supplementary Table S8). Further enrichment analyses revealed the ceRNA network-related signaling pathways, including “synaptic vesicle cycle,” “Fc gamma R-mediated phagocytosis,” and “positive regulation of JUN kinase activity” (Fig. 5C, D; Supplementary Table S10). The GO enrichment analysis of key ceRNA networks revealed that syntaxin-binding protein 1 ( STXBP1 ) and secretory carrier membrane protein 5 ( SCAMP5 ) are involved in the positive regulation of calcium-dependent exocytosis. And tyrosine kinase ephrin receptor A4 ( EPHA4 ), protein kinase C epsilon ( PRKCE ), and STXBP1 are jointly involved in the regulation of transsynaptic signaling (Supplementary Table S10). Validation of ceRNA networks We assessed the role of lncRNA dysregulation in altered mRNA levels. In the ceRNA network, MIR600HG and AL049775.1 had strong correlations with 11 DEmRNAs, as assessed by Pearson’s correlation analysis (correlation coefficients > 0.75) (Supplementary Fig. S4; Supplementary Table S11). Of these, MIR600HG and MAP3K9 had the strongest correlation with high correlation coefficients at 0.91. Furthermore, STXBP1 has a positive correlation with another mRNA (correlation coefficients > 0.85). For example, STXBP1 had interacts with MAP3KP9 (r = 0.93), SCAMP5 (r = 0.94), PRKCE (r = 0.94), EPHA4 (r = 0.86), and MOAP1 (r = 0.94). In addition, four microarrays and RNA-seq datasets used for validation were integrated, and the downregulated expression of marker genes from ceRNA networks was observed in the autistic brain tissue (Fig. 5F). MIR600HG was found to be downregulated in GSE30573 (log2FC = −2.01) and GSE64018 (log2FC = −0.49) (Fig. 5F and Supplementary Table S12). Validation of the hub genes in clinical samples MIR600HG is the most closely related to nine known autism-susceptible miRNAs (Fig. 5B). Among the identified lncRNA-miRNA-mRNA interactions, we selected a DE-lncRNA ( MIR600HG , which was downregulated in multiple datasets) for the following experimental validations. MIR600HG exhibited significant differential expression between patients with autism and controls by qRT-PCR (log2FC = −0.62, FDR = 2.44E-04) (Fig. 5E and Supplementary Table S13), and differences were observed in the male samples (log2FC = −0.64, FDR = 1.81E-03) instead of the females (Supplementary Fig. S5). PAK1 was downregulated in the postmortem cortex and upregulated in whole blood. However, 6/10 DE-mRNAs ( SLC12A5 excluded) were significantly downregulated, including EPHA4 (log2FC = −0.56, FDR = 1.45E-05), MAP3K9 (log2FC = −0.47, FDR = 7.81E-07), MOAP1 (log2FC = −0.51, FDR = 1.14E-10), STXBP1 (log2FC = −0.38, FDR = 2.28E-03), PRKCE (log2FC = −0.54, FDR = 1.95E-08), and SCAMP5 (log2FC = −0.54, FDR = 5.06E-05). The relative expressions of these mRNAs, except for SCAMP5 , were significantly downregulated in the peripheral blood of both male and female patients (Supplementary Table S13; Supplementary Fig. S5). Concurrently, the prefrontal cortex samples obtained from male patients exhibited a significant downregulation in the expression levels of MIR600HG and its associated target genes. In contrast, the female samples displayed a declining trend, but the statistical analysis did not yield significant evidence to support this observation (Supplementary Fig. S6). Correlation verification of mRNA regulation by MIR600HG We found that significant DE-lncRNA ( MIR600HG ) and six differential mRNAs were downregulated in the brain tissue and peripheral blood. We observed an upregulation trend in nine specific miRNAs following interference with MIR600HG . Notably, hsa-miR-148b-3p, hsa-miR-7-5p, hsa-miR-106b-5p, and hsa-miR-21-5p were significantly upregulated with statistical significance (Supplementary Fig. S7). Previous studies have reported upregulation of these miRNAs in the brain tissue of individuals with ASD (Supplementary Table S9). These findings provide further support for the regulatory role of MIR600HG in modulating the expression of these specific miRNAs. The results showed that MIR600HG knockdown affects the expression of its corresponding mRNAs ( STXBP1 , MAP3K9 , EPHA4 , and MOAP1 ) (Fig. 5G). Notably, we examined the expression of MIR600HG and the mRNA counterparts in various human tissues by GTEx profiles and found that these genes were enriched in the brain (Supplementary Fig. S8A). RNA-seq data from the BrainSpan project showed that MIR600HG is broadly expressed in brain regions, but developmentally regulated in the human brain from pregnancy to adulthood. In this study, six main brain regions (dorsolateral prefrontal cortex (DFC); ventrolateral prefrontal cortex (VFC); medial prefrontal cortex (MFC); orbital frontal cortex (OFC); hippocampus (HIP), and inferolateral temporal cortex (ITC)) were investigated. MIR600HG has spatiotemporal co-expression with the targeted mRNA at each developmental stage (prenatal, postnatal, and adulthood), which may have similar molecular pathways or functions. The expression patterns of MIR600HG and mRNA were similar in different human brain regions and at different stages of development (Supplementary Fig. S8B–G). Together, these findings hinted at the role of a new lncRNA MIR600HG in regulating ASD-associated miRNAs and mRNAs, suggesting a critical role of the ceRNA network in ASD. Construction of a diagnostic model based on machine learning algorithms Machine learning algorithms were selected for the diagnostic modeling of the marker genes of ASD in the ceRNA network. First, SVM-RFE analysis revealed that the first 10 genes were identified as potential genes based on an optimum error rate (0.316). Similarly, the RF algorithm screened 11 ASD-associated diagnostic marker genes (Supplementary Fig. S9A). Finally, the common genes obtained by the above two algorithms, PRKCE , CALM3 , STXBP1 , GRAMD1B , MAP3K9 , PAK1 , MOAP1 , KIAA0513 , and MIR600HG , were utilized to construct a diagnostic nomogram (Supplementary Fig. S9B). The analyses of the AUC revealed that these 9 genes hold potential as diagnostic biomarkers. The AUC of the diagnostic nomogram was 0.851 for prefrontal cortex samples, and the diagnostic performance of the model was satisfactory (AUC > 0.65) in peripheral blood samples (Supplementary Fig. S9C–E). Discussion The lncRNA and mRNA co-expression module was obtained by WGCNA analysis and detected along with the DE genes for the enrichment of autism-related gene signals. The ceRNA network was constructed by integrating the candidate miRNA gene sets from the literature. RT-PCR assays confirmed that lncRNAs and target genes are dysregulated at the peripheral circulatory level. Next, we hypothesized that MIR600HG regulating transcription-level genes through the mechanism of ceRNA may lead to ASD susceptibility. This model is supported by a positive correlation between the expression of lncRNAs and mRNAs affected by ASD; also, the regulation of miRNA and mRNA targets was assessed by lncRNA knockdown in cells. Overall, our findings suggested that ASD-associated transcriptomic changes may be partially attributed to lncRNA dysregulation. MIR600HG is a common intersection of DGE analysis and co-expression M4 modules. WGCNA analysis determined that M4 modules (downregulated) are correlated with ASD and significantly overlapped with PSD proteins and synaptic signaling function. Furthermore, the function of upregulated genes and modules was related to immunity, as described previously [33, 34]. However, the upregulated genes did not show an enrichment of the genetic components. Previous studies indicated that lncRNA is a transcriptional and post-transcriptional regulator, and dysregulation of ncRNAs plays a critical role in the pathogenesis of ASD [6, 7, 11, 13]. MIR600HG in Alzheimer’s disease regulates Aβ accumulation [35] and is associated with tumors [36, 37]. In the present study, MIR600HG was downregulated in multiple datasets of postmortem brain tissue. The dysregulation of MIR600HG expression and the role of the ceRNA regulatory axis in ASD have never been reported. Also, we highlighted that MIR600HG interacts with multiple mRNAs through autism-susceptible miRNAs (for example, hsa-miR-106a/b-5p, hsa-miR-107, hsa-miR-92a-3p, hsa-miR-15b-5p, hsa-miR-21-5p, and hsa-miR-148b-3p). The miRNA of interest has been associated with ASD, providing a useful set of upstream regulatory (lncRNA-miRNA axis) target genes. In this study, it was found that the expression of some miRNAs was significantly upregulated after inhibiting the expression of MIR600HG using shRNA knockdown. These findings further support the role of lncRNA as endogenous “sponges” for miRNAs [15]. These dysregulated miRNAs might affect the expression of genes related to autism and neurodevelopment [38,39,40,41]. The target mRNAs were downregulated in the brain tissue (GEO datasets) and peripheral blood (Han Chinese populations), including STXBP1 , EPHA4 , MAP3K9 , MOAP1 , PRKCE , and SCAMP5 . STXBP1 , localized primarily in the cell body and axon, is associated with vesicle fusion and neurotransmitter release throughout development [42,43,44]. The inactivation of Munc18-1 in mice leads to widespread neurodegeneration [43] and synaptic impairments [45]. A decrease in STXBP1 by patient-derived induced pluripotent stem cells (iPSCs) to generate induced neurons resulted in neurite extension defects [46]. The mutations in STXBP1 are associated with neurodevelopmental disorders, such as ASD, developmental disorders, and epileptic encephalopathy [47,48,49]. Recurrent de novo and likely gene-disruptive mutations for STXBP1 have been reported in a Chinese ASD patient cohort [50]. SCAMP5 is highly expressed in the brain and is involved in transmitting nerve signals, regulating axonal trafficking, synaptic localization, and synaptic plasticity [51]. De novo SCAMP5 mutation causes a neurodevelopmental disorder with autistic features [52]. SCAMP5 has been shown to interact with soluble N-ethylmaleimide sensitive factor attachment protein receptors (SNAREs) molecules, which are important for intracellular vesicular trafficking and developmental psychiatry [53, 54]. EPHA4 belongs to the A subgroup of Eph receptors, which are key players in synaptic plasticity and neural development [55, 56]. Rare changes in EPHA4/p.P775L are linked to ASD [48] and EPHA3 identified as candidate genes in ASD [57]. Both EphA4 and EphA7 affect cortical neuronal migration during mouse brain development [56]. EphA4 knock-out (-/-) mice displayed impaired movements, which are associated with disruption of axon-guided function [58]. MAP3K9 regulates signaling by the mitogen-activated protein kinase (MAPK) and c-Jun amino-terminal kinase (JNK) pathways [59]. The MAPK signaling pathway, critical for neurodevelopment, is involved in neurogenesis migration and the development of dendritic trees and spines [60]. Some studies have shown that the MAPK signaling pathway and mitochondrial dysfunction are involved in the pathogenesis of ASD [61]. Furthermore, in the present study, among the ceRNA network, MIR600HG had a strong correlation with DEmRNAs. LncRNAs regulate gene expression in multiple ways at the epigenetic, chromatin remodeling, transcription, and translation levels, and as ceRNAs that attenuate the role of miRNAs on targeted messenger RNA (mRNA) involved in the development of tumors and neurological diseases [5, 10, 15, 62, 63]. Recently, the mechanisms of immune-related ceRNA regulation in ASD diseases have been deduced [64, 65]. However, the role of the synaptic-associated ceRNA regulatory axis in ASD has never been reported and remains largely unknown. In the present study, we provided the corresponding ASD-associated ceRNA network and characterized the targets of MIR600HG -downregulated mRNA axis (for example, hsa-miR-21-5p/hsa-miR-106b-5p- EPHA4 and hsa-miR-148b-3p- MAP3K9 axis) in vitro. Thus, elucidating specific spatial and temporal expression patterns of lncRNAs is crucial to identifying the role of lncRNAs in nervous system development [66]. In this study, the expression patterns of MIR600HG and targeted mRNA were similar in different human brain regions and at different stages of development, suggesting that they may have similar molecular pathways or functions. Another study reported that the JNK and MAP kinase pathways play a crucial role in synaptic formation [67]. Interestingly, functional analysis revealed that EPHA4 and MAP3K9 genes are related to JUN kinase activation, and EPHA4 and STXBP1 are enriched in synaptic signal transduction and regulation of calcium ion-dependent exocytosis; all these are involved in the pathogenesis of ASD. Moreover, lncRNAs, such as metastasis-associated lung adenocarcinoma transcript 1 (MALAT1 ) [68], BDNF-AS [69], brain cytoplasmic RNA 1 ( BCYRN1 ) [70] regulate synapsis and synaptic plasticity through alternative splicing or the expression of synaptic-related genes. In summary, MIR600HG potentially regulates the expression of synaptic function-related genes at the transcription level through ceRNA mechanisms. Nevertheless, the present study has several limitations. For example, further functional exploration of lncRNAs and mRNAs with spatiotemporal expression pattern consistency is essential. Due to the species-specific expression of this particular lncRNA ( MIR600HG ) in humans, future investigations should prioritize the inclusion of appropriate animal models to validate and extend the findings of this study. The young brain samples from patients and patient-derived brain organoids that mimic early brain development will help in characterizing the dysregulation of lncRNAs and potential ceRNA regulation mechanisms in autism. In the future, the investigation of the manipulation of MIR600HG expression in the brain and studying its impact at the molecular and behavioral levels would provide a more comprehensive exploration of the functional significance of this lncRNA in brain development and behavior. Moreover, the present study focuses on transcriptomics because the transcription process is affected by many factors, and mRNA changes may vary between the blood and the brain. Thus, the current findings need to be replicated with larger samples and different ethnic backgrounds. The limited sample size of female ASD patients in our study may have contributed to the lack of statistical significance in the observed declining trend of MIR600H G expression and its associated target genes. Future studies with larger cohorts of female ASD patients are required to validate and further explore these observations. In conclusion, dysregulation of MIR600HG potentially regulates the expression of synaptic function-related genes at the transcription level through ceRNA mechanisms. Consequently, our findings corroborate the role of lncRNA dysregulation and synaptic signaling-related ceRNA regulatory axis in ASD. Our comprehensive bioinformatics analysis provides a framework for assessing the functional participation of lncRNAs in ASD. The role of lncRNA in the development and function of the central nervous system needs to be investigated further. Data availability The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors. References Geschwind DH, State MW. Gene hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol. 2015;14:1109–20. Geschwind DH, Flint J. Genetics and genomics of psychiatric disease. Science. 2015;349:1489–94. Hosseini E, Bagheri-Hosseinabadi Z, De Toma I, Jafarisani M, Sadeghi I. The importance of long non-coding RNAs in neuropsychiatric disorders. Mol Asp Med. 2019;70:127–40. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–89. Tang J, Yu Y, Yang W. Long noncoding RNA and its contribution to autism spectrum disorders. CNS Neurosci Ther. 2017;23:645–56. Ng SY, Johnson R, Stanton LW. Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. 2012;31:522–33. Ng SY, Bogu GK, Soh BS, Stanton LW. The long noncoding RNA RMST interacts with SOX2 to regulate neurogenesis. Mol Cell. 2013;51:349–59. Neuhaus E, Beauchaine TP, Bernier R. Neurobiological correlates of social functioning in autism. Clin Psychol Rev. 2010;30:733–48. Young AM, Chakrabarti B, Roberts D, Lai MC, Suckling J, Baron-Cohen S. From molecules to neural morphology: understanding neuroinflammation in autism spectrum condition. Mol Autism. 2016;7:9. Fang Y, Fullwood MJ. Roles, functions, and mechanisms of long non-coding RNAs in cancer. Genomics Proteomics Bioinformatics. 2016;14:42–54. Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature. 2016;540:423–7. Ziats MN, Rennert OM. Aberrant expression of long noncoding RNAs in autistic brain. J Mol Neurosci. 2013;49:589–93. Bond AM, Vangompel MJ, Sametsky EA, Clark MF, Savage JC, Disterhoft JF, et al. Balanced gene regulation by an embryonic brain ncRNA is critical for adult hippocampal GABA circuitry. Nat Neurosci. 2009;12:1020–7. Wang Y, Zhao X, Ju W, Flory M, Zhong J, Jiang S, et al. Genome-wide differential expression of synaptic long noncoding RNAs in autism spectrum disorder. Transl Psychiatry. 2015;5:e660. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344–52. Liu X, Han D, Somel M, Jiang X, Hu H, Guijarro P, et al. Disruption of an evolutionarily novel synaptic expression pattern in autism. PLoS Biol. 2016;14:e1002558. Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7. Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell. 2013;155:1008–21. Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–21. Werling DM, Parikshak NN, Geschwind DH. Gene expression in human brain implicates sexually dimorphic pathways in autism spectrum disorders. Nat Commun. 2016;7:10717. Zeisel A, Munoz-Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, Jureus A, et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347:1138–42. Bayes A, van de Lagemaat LN, Collins MO, Croning MD, Whittle IR, Choudhary JS, et al. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat Neurosci. 2011;14:19–21. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362–8. Huang HY, Lin YC, Li J, Huang KY, Shrestha S, Hong HC, et al. miRTarBase 2020: updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 2020;48:D148–54. Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48:D127–31. Rawat M, Nighot M, Al-Sadi R, Gupta Y, Viszwapriya D, Yochum G, et al. IL1B increases intestinal tight junction permeability by up-regulation of MIR200C-3p, which degrades occludin mRNA. Gastroenterology. 2020;159:1375–89. Wang Z, Lu T, Li X, Jiang M, Jia M, Liu J, et al. Altered expression of brain-specific autism-associated miRNAs in the Han Chinese population. Front Genet. 2022;13:865881. Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1826. Huang ML, Hung YH, Lee WM, Li RK, Jiang BR. SVM-RFE based feature selection and Taguchi parameters optimization for multiclass SVM classifier. ScientificWorldJournal. 2014;2014:795624. Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57:67–81. Meltzer A, Van de Water J. The role of the immune system in autism spectrum disorder. Neuropsychopharmacology. 2017;42:284–98. Liu Q, Ling Z, Zhang J, Yu H, Wang Y, Xue Y, et al. lncRNA MIR600HG knockdown alleviates cognitive impairment in Alzheimer’s disease through NEDD4L mediated PINK1 degradation. J Alzheimers Dis. 2022;85:1783–94. Xiao J, Lv C, Xiao C, Ma J, Liao J, Liu T, et al. Construction of a ceRNA network and analysis of tumor immune infiltration in pancreatic adenocarcinoma. Front Mol Biosci. 2021;8:745409. Liu X, Zhao T, Yuan Z, Ge S. MIR600HG sponges miR-125a-5p to regulate glycometabolism and cisplatin resistance of oral squamous cell carcinoma cells via mediating RNF44. Cell Death Discov. 2022;8:216. Abu-Elneel K, Liu T, Gazzaniga FS, Nishimura Y, Wall DP, Geschwind DH, et al. Heterogeneous dysregulation of microRNAs across the autism spectrum. Neurogenetics. 2008;9:153–61. Sarachana T, Zhou R, Chen G, Manji HK, Hu VW. Investigation of post-transcriptional gene regulatory networks associated with autism spectrum disorders by microRNA expression profiling of lymphoblastoid cell lines. Genome Med. 2010;2:23. Wu YE, Parikshak NN, Belgard TG, Geschwind DH. Genome-wide, integrative analysis implicates microRNA dysregulation in autism spectrum disorder. Nat Neurosci. 2016;19:1463–76. Nguyen LS, Lepleux M, Makhlouf M, Martin C, Fregeac J, Siquier-Pernet K, et al. Profiling olfactory stem cells from living patients identifies miRNAs relevant for autism pathophysiology. Mol Autism. 2016;7:1. Rizo J, Sudhof TC. The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices–guilty as charged? Annu Rev Cell Dev Biol. 2012;28:279–308. Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, et al. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–9. Cijsouw T, Weber JP, Broeke JH, Broek JA, Schut D, Kroon T, et al. Munc18-1 redistributes in nerve terminals in an activity- and PKC-dependent manner. J Cell Biol. 2014;204:759–75. Miyamoto H, Shimohata A, Abe M, Abe T, Mazaki E, Amano K, et al. Potentiation of excitatory synaptic transmission ameliorates aggression in mice with Stxbp1 haploinsufficiency. Hum Mol Genet. 2017;26:4961–74. Yamashita S, Chiyonobu T, Yoshida M, Maeda H, Zuiki M, Kidowaki S, et al. Mislocalization of syntaxin-1 and impaired neurite growth observed in a human iPSC model for STXBP1-related epileptic encephalopathy. Epilepsia. 2016;57:e81–6. Zhang Y, Wang R, Liu Z, Jiang S, Du L, Qiu K, et al. Distinct genetic patterns of shared and unique genes across four neurodevelopmental disorders. Am J Med Genet B Neuropsychiatr Genet. 2021;186:3–15. Marchese M, Valvo G, Moro F, Sicca F, Santorelli FM. Targeted gene resequencing (Astrochip) to explore the tripartite synapse in autism-epilepsy phenotype with macrocephaly. Neuromol Med. 2016;18:69–80. Stamberger H, Nikanorova M, Willemsen MH, Accorsi P, Angriman M, Baier H, et al. STXBP1 encephalopathy: a neurodevelopmental disorder including epilepsy. Neurology. 2016;86:954–62. Wang T, Guo H, Xiong B, Stessman HA, Wu H, Coe BP, et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat Commun. 2016;7:13316. Lee U, Choi C, Ryu SH, Park D, Lee SE, Kim K, et al. SCAMP5 plays a critical role in axonal trafficking and synaptic localization of NHE6 to adjust quantal size at glutamatergic synapses. Proc Natl Acad Sci USA. 2021;118:e2011371118. Hubert L, Cannata Serio M, Villoing-Gaude L, Boddaert N, Kaminska A, Rio M, et al. De novo SCAMP5 mutation causes a neurodevelopmental disorder with autistic features and seizures. J Med Genet. 2020;57:138–44. Han C, Chen T, Yang M, Li N, Liu H, Cao X. Human SCAMP5, a novel secretory carrier membrane protein, facilitates calcium-triggered cytokine secretion by interaction with SNARE machinery. J Immunol. 2009;182:2986–96. Cupertino RB, Kappel DB, Bandeira CE, Schuch JB, da Silva BS, Muller D, et al. SNARE complex in developmental psychiatry: neurotransmitter exocytosis and beyond. J Neural Transm. 2016;123:867–83. Vargas LM, Cerpa W, Munoz FJ, Zanlungo S, Alvarez AR. Amyloid-beta oligomers synaptotoxicity: the emerging role of EphA4/c-Abl signaling in Alzheimer’s disease. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1148–59. Son AI, Hashimoto-Torii K, Rakic P, Levitt P, Torii M. EphA4 has distinct functionality from EphA7 in the corticothalamic system during mouse brain development. J Comp Neurol. 2016;524:2080–92. Casey JP, Magalhaes T, Conroy JM, Regan R, Shah N, Anney R, et al. A novel approach of homozygous haplotype sharing identifies candidate genes in autism spectrum disorder. Hum Genet. 2012;131:565–79. Jiang J, Kullander K, Alstermark B. EphA4 is required for neural circuits controlling skilled reaching. J Neurosci. 2020;40:7091–104. Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat Rev Mol Cell Biol. 2002;3:663–72. Albert-Gasco H, Ros-Bernal F, Castillo-Gomez E, Olucha-Bordonau FE. MAP/ERK signaling in developing cognitive and emotional function and its effect on pathological and neurodegenerative processes. Int J Mol Sci. 2020;21:4471. Yang J, He X, Qian L, Zhao B, Fan Y, Gao F, et al. Association between plasma proteome and childhood neurodevelopmental disorders: a two-sample Mendelian randomization analysis. EBioMedicine. 2022;78:103948. Wang W, Min L, Qiu X, Wu X, Liu C, Ma J, et al. Biological function of long non-coding RNA (LncRNA) Xist. Front Cell Dev Biol. 2021;9:645647. Ma N, Tie C, Yu B, Zhang W, Wan J. Identifying lncRNA-miRNA-mRNA networks to investigate Alzheimer’s disease pathogenesis and therapy strategy. Aging. 2020;12:2897–920. Sun JJ, Chen B, Yu T. Construction of an immune-related ceRNA network to screen for potential diagnostic markers for autism spectrum disorder. Front Genet. 2022;13:1025813. Sabaie H, Dehghani H, Shiva S, Asadi MR, Rezaei O, Taheri M, et al. Mechanistic insight into the regulation of immune-related genes expression in autism spectrum disorder. Front Mol Biosci. 2021;8:754296. Zhang XQ, Wang ZL, Poon MW, Yang JH. Spatial-temporal transcriptional dynamics of long non-coding RNAs in human brain. Hum Mol Genet. 2017;26:3202–11. Jiang X, Sando R, Sudhof TC. Multiple signaling pathways are essential for synapse formation induced by synaptic adhesion molecules. Proc Natl Acad Sci USA. 2021;118:e2000173118. Bernard D, Prasanth KV, Tripathi V, Colasse S, Nakamura T, Xuan Z, et al. A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J. 2010;29:3082–93. Modarresi F, Faghihi MA, Lopez-Toledano MA, Fatemi RP, Magistri M, Brothers SP, et al. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat Biotechnol. 2012;30:453–9. Muslimov IA, Santi E, Homel P, Perini S, Higgins D, Tiedge H. RNA transport in dendrites: a cis-acting targeting element is contained within neuronal BC1 RNA. J Neurosci. 1997;17:4722–33. Funding This work was supported by grants from the Key-Area Research and Development Program of Guangdong Province (2019B030335001), the National Natural Science Foundation of China (grant numbers 81971283, 82171537, 82071541, 81671363, and 82271576), the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2023-PT320-08) and the Young Elite Scientists Sponsorship Program by CAST (YESS20160068). Finally, we would also like to thank all the participants and their families. Ethics declarations Competing interests The authors declare no competing interests. Ethics approval The studies involving human participants were reviewed and approved by the ethics committee of Peking University Sixth Hospital (Beijing, China). Informed consent Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Additional information Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Supplementary information Rights and permissions Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. About this article Cite this article Jiang, M., Wang, Z., Lu, T. et al. Integrative analysis of long noncoding RNAs dysregulation and synapse-associated ceRNA regulatory axes in autism.
Transl Psychiatry 13 , 375 (2023). https://doi.org/10.1038/s41398-023-02662-5 Received: Revised: Accepted: Published: DOI: https://doi.org/10.1038/s41398-023-02662-5
This content was originally published here.